()
Catalog#:
Size:
Instruction:


 Gene Editing Services
Gene Editing Services
Ubigene has modified over 5000 genes from more than 200 cell lines with our exclusive innovation CRISPR-U™ technology. At the same time, we already provide customers with high quality gene-editing tools for cell or animal research worldwide.
 EZ-editor™
EZ-editor™With 14 years of experience, Ubigene has exclusively innovated and developed 6 product lines, fullfilling all kinds of needs from researchers. Experiment process simplified, efficiency improved, achieving our aim of 'Make genome editing easier'!
 Red Cotton Gene knockout Project
Red Cotton Gene knockout Project
Ubigene exclusive KO Cell Line Bank, over 5000 KO cell lines, covering thousands of genes from 8 popular signaling pathways and nearly 100 diseases.
Location:Home > Application > Knockout cell pool boost researcher confidence in gene function study
Knockout cell pool boost researcher confidence in gene function study
CRISPR is a revolutionary genome-editing technology and it can be used to edit genes and, as such, will likely change the world. Although it’s a relatively recent discovery, CRISPR quickly becomes a standard laboratory tool in biological research. One of the most widely used applications of CRISPR is knocking out specific genes in cell lines to interrogate gene function. The experimental setup often requires transfecting a large number of cells at once, resulting in a heterogeneous pool of edited cells with varying indel population termed as Knockout (KO) pools. CRISPR generated KO cell pools are becoming the method of choice for many researchers investigating gene function in cell lines, replacing the older RNAi-based approaches. However, the gene knockout does not impact cell viability, and the total population of edited cells does not change over time. The KO cell pools contain a mix of edited and wild type cells, the knockout efficiency may vary over time.KO cell pool is highly useful for short term assay.
CRISPR generated cell Pools with high KO frequencies are suitable for direct use in many types of assays, without a requirement for first generating clonal cell lines. A genotypic aberration can directly lead to an observable phenotypic effect. Knockout cell pools have wide application in the following research area.
1. Proliferation assay
2. Analyze signaling pathway involvement
3. Drug response/ Drug target identification
4. Disease Model developments
5. Antibody validation (Immunoblot/ Immunofluorescence)
The CRISPR system, a potent system for genome editing, is used for gene knockout or knock-in genome manipulations through the substitution of a target genetic sequence with a desired donor sequence. CRISPR technology allows us to generate knockout cell pools as a polyclonal population of cells, rather than having to take the time and effort to go clonal, which needed more time and increases the cost. Nowadays, scientists start to switch RNAi to using knockout cell pools for their early exploratory studies because the knockout cell pools are that they lower that barrier to entry and are very similar inaccessibility to RNAi. Moreover, CRISPR can generate a stable knockout of protein function which can provide cleaner results than RNAi. Knockout cell pool with higher efficiency is very useful for the study of drug targets, drug response, antibody validation, disease model development as well as various functional assay studies. To accelerate biomedical research, Ubigene developed CRISPR-U™ for gene manipulated knockout cell pool generation. The CRISPR-U™ system is used for rapid and precise gene editing for knockout cell generation. Besides, CRISPR-generated cell pools could be coupled with a loss of function assays, high throughput screening, antibody validation, and safety pharmacology studies. Ubigene can customize the gene-editing knockout cell line and cell pool generation as well as can generate various genes modification in animal models.
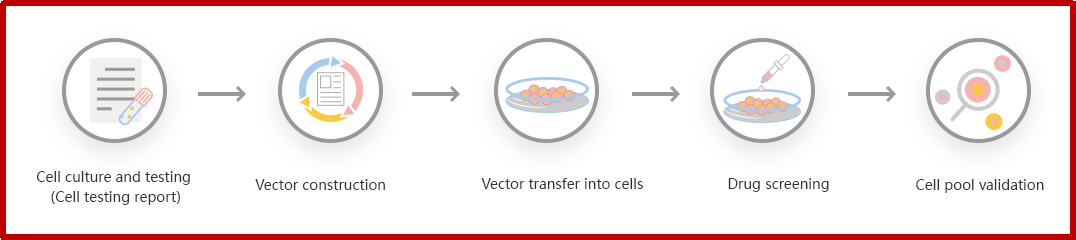
Figure: CRISPR-U™ customized workflow for engineered KO cell pool
Ubigene developed CRISPR-U™ which optimizes KO cell pool gene editing. The efficiency and accuracy are higher than traditional methods. Contact us immediately to know about your research related services!
Knockout cell pool for low or undetectable residual target proteins expression study
CRISPR/Cas9-based gene knockouts (KOs) enable precise perturbation of target gene function and these phenomena are ideally assessed in an unbiased fashion by molecular omics readouts in human cells. Typically, this requires the lengthy process of isolating KO subclones. In this study, the researcher used a strategy that avoids subcloning and achieves fast and efficient gene silencing on cell pools, based on the synergistic combination of two guide RNAs mapping at close (40-300 bp) genomic proximity. The researchers, knockout pyridoxal kinase (PDXK) in HepG2 cells and generate KO PDXK cell pools. They also generate KO clones of PDXK. They compared the phenotypic proteomics data generated in KO clones as well as KO cell pools.
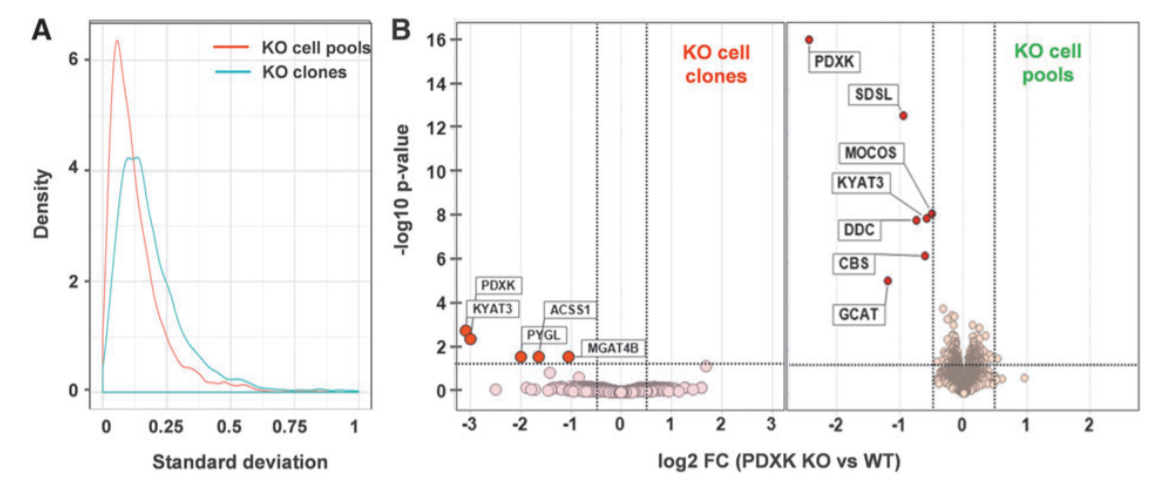
Figur1. Proteomics phenotype of PDXK KO cells.
The standard deviation between replicates shows that the cell pool dataset shows less variation around the mean than the dataset from the three PDXK KO clones confirming the heterogeneity (A). An unbiased whole proteome analysis identified six proteins that significantly down-regulated HepG2 KO cell pool, whereas in KO clones four proteins were significantly down-regulated. MS quantification of the residual levels of the cognate protein is required to exclude that truncated versions of the protein are expressed following an early stop codon or exon skipping which might confound the analysis and interpretation of the KO phenotype.
Therefore, KO cell pool is highly useful for investigating gene function in cell lines, replacing the older RNAi-based approaches.
Reference:
A Tandem Guide RNA-Based Strategy for Efficient CRISPR Gene Editing of Cell Populations with Low Heterogeneity of Edited Alleles. CRISPR J.2020;3(2):123-134.





 Back To Top
Back To Top
Our Solution Specialists will contact you as soon as possible!