()
Catalog#:
Size:
Instruction:


 Gene Editing Services
Gene Editing Services
Ubigene has modified over 6000 genes from more than 300 cell lines with our exclusive innovation CRISPR-U™ technology. At the same time, we already provide customers with high quality gene-editing tools for cell or animal research worldwide.
 EZ-editor™
EZ-editor™With 15 years of experience, Ubigene has exclusively innovated and developed 6 product lines, fullfilling all kinds of needs from researchers. Experiment process simplified, efficiency improved, achieving our aim of 'Make genome editing easier'!
 Red Cotton Gene knockout Project
Red Cotton Gene knockout Project
Ubigene exclusive KO Cell Line Bank, over 5000 KO cell lines, covering thousands of genes from 8 popular signaling pathways and nearly 100 diseases.
Location:Home > Application > ARPE-19, Genome Surgery and Vision Science, A combo for tackling blindness
ARPE-19, Genome Surgery and Vision Science, A combo for tackling blindness

ARPE-19 is a spontaneously arising retinal pigment epithelial (RPE) cell line derived in 1986 by Amy Aotaki-Keen from the normal eyes of a 19-year-old male, who died from head trauma in a motor vehicle accident. These cells form a stable monolayer, which exhibits morphological and functional polarity. ARPE-19 cell line has a visually normal karyotype and expresses RPE-specific markers, the retinal pigment epithelium-specific 65kDa protein (RPE65), and cellular retinaldehyde-binding protein (CRALBP), as has been shown at the mRNA and protein levels, respectively.
ARPE-19 is one of the popular cellular models as well as tools for vision scientists, who are willing to study the occurrence of ophthalmic disorders, mechanism of action of diseases, or therapeutically active drug molecules as well as decipher cell signaling events. Retinal disorders can cause impaired vision, severe vision loss or even blindness. Genetic alterations resulting in a dysfunctional retinal pigment epithelium and/or degenerating photoreceptors cause impaired vision. These juxtaposed cells in the retina of the posterior eye are crucial for the visual cycle or phototransduction. Deficits in these biochemical processes perturb neural processing of images capturing the external environment.
To date, there are more than 260 different genes were identified that cause inherited retinal disorders (IRDs). Each IRD is caused by at least one gene that is not working as it should. IRDs can affect individuals of all ages, can progress at different rate, and are rare. However, many are degenerative, which means that the symptoms of the disease will get worse over time. Common types of IRDs include Leber Congenital Amaurosis (LCA), Retinitis Pigmentosa, Choroideremia, Stargardt’s Disease, and Achromatopsia, etc. Notably, there is a distinct lack of clinically approved pharmacological, cell- or gene-based therapies for inherited retinal disease.
Gene editing technologies are rapidly advancing as a realistic therapeutic option. The ability to strategically edit a patient’s genome can constitute a treatment revolution. However, concerns remain over the safety and efficacy of either transplanting retinal cells following ex vivo gene editing, or with direct gene editing in vivo. Ultimately, further refinements to improve efficacy and safety profiles are paramount for gene editing to emerge as a widely available treatment option.
ARPE-19 cell lines application in Vision science.
1. Basic Research: Understanding cell fate control, Studying pluripotency, study retinal cell biology, pathophysiology and pharmacology.
2. Drug Discovery: The most widely used RPE model in ocular drug discovery and drug screening. Drug discovery for diabetic retinopathy (DR), age-related macular degeneration (AMD), and retinitis pigmentosa, etc.
3. Toxicology Studies: Pharmacokinetics at cellular level, studied uptake of drugs and toxicity testing.
4. Disease Modeling: A useful and convenient model for Diabetic Retinopathy (DR), Macula edema research, age-related macular degeneration (AMD), and retinitis pigmentosa.
Clustered regularly interspaced short palindromic repeats (CRISPR)-Cas9 technology is currently the leading gene-editing technology positioned for therapeutic intervention. Genome editing and the creation of cellular models for ophthalmology can advance research programs in the area of functional genomics, signaling pathways, metabolism, cell death, drug discovery, drug response and cell therapy research, etc. Furthermore, gene editing is emerging as an attractive novel treatment strategy for inherited retinal disease.
Recently, CRISPR/Cas9 was applied to ex-vivo correction of an X-linked retinitis pigmentosa GTPase regulator (RPGR) inherited retinal disorders mutation (IRD) and corrected c.3070G>T mutation. This landmark treatment uses the CRISPR approach to a specific mutation in a gene linked to childhood blindness. This proof-of-concept study demonstrates the capability to repair genes and a treatment milestone can be achieved if these findings are safely and effectively applied to patient eyes. To accelerate ophthalmology and visual sciences research, Ubigene developed CRISPR-U™ for gene manipulation of eukaryotic cells. Thereby, possible to achieve genome editing in APRE-19 cells with the utilization of the CRISPR/Cas9 system. The CRISPR-U™ system is used for rapid and precise gene editing in APRE-19 cells. CRISPR-generated cell models have enabled mechanism explorations of various visually impairs diseases, which are mainly related to retinal pathogenesis, toxicity, polarity studies of proteins, gene delivery, multi-omics, drug screening, and signaling pathways.
Due to the unique features of the human eye and complexities associated with the nature of the disease, animal models fail to mimic all aspects of physiology and pathology of eye diseases. Therefore, cell culture models are useful alternative tools to investigate the physiology and pathology of diseases. Besides, cell culture models are advantageous because they are experimentally controlled systems and so the results are more reproducible than those obtained from animal models. Based on the elucidation of disease mechanisms, CRISPR-generated cell models could be coupled with drug discovery, drug screening and other therapeutic studies. Ubigene can customize the gene-editing in eukaryotic cells as well as can generate various genes modification in animal models.
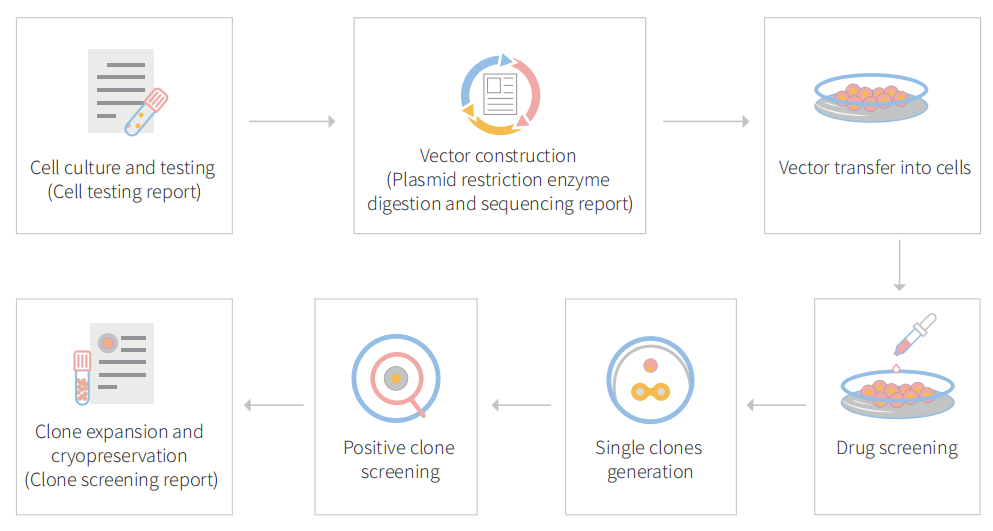
Figure: CRISPR-U™ customized workflow for engineered APRE-19 model cells
Prominin-1 act as a regulator in human retinal pigment epithelium
Prominin-1 (Prom1) is a trans-membrane glycoprotein, which is expressed in stem cell lineages, and have a role in cancer stem cell survival. In the visual system, Prom1 is concentrated in the photoreceptor outer segment disc membranes and is thought to play a structural role. The expression of the human dominant Prom1 R373C mutation in mice disrupted photoreceptor disk morphogenesis, suggesting that Prom1 plays an integral role in the structural organization of the outer segment. Also, retinal degeneration has been reported in a spontaneous knock out mouse (Prom1rd19) carrying a point mutation in the Prom1 gene. Human mutations in the Prom1 gene cause Stargardt like and bull’s eye macular dystrophies and retinitis pigmentosa. One critical function performed by the retinal pigments epithelium (RPE) is the phagocytosis and lysosomal degradation of shed photoreceptor outer segment tips. Recent studies have shown that autophagy by the RPE is fundamental to this activity. In this study, the researcher investigates the role of prominin1 in metabolically active cells of retinal pigments epithelium (RPE).
The researcher investigated the functional role and significance of Prom1 in the RPE by using a lentiviral construct overexpressing Prom1 and CRISPR/Cas9 knockout (KO) of Prom1 in the RPE. They demonstrate that Prom1 is a novel and key regulator of autophagosome formation and turnover. Moreover, they reveal a molecular mechanism by which Prom1 regulates autophagy flux in the RPE via p62 and HDAC6 association, and by its ability to inhibit mTORC1 and mTORC2 activities.
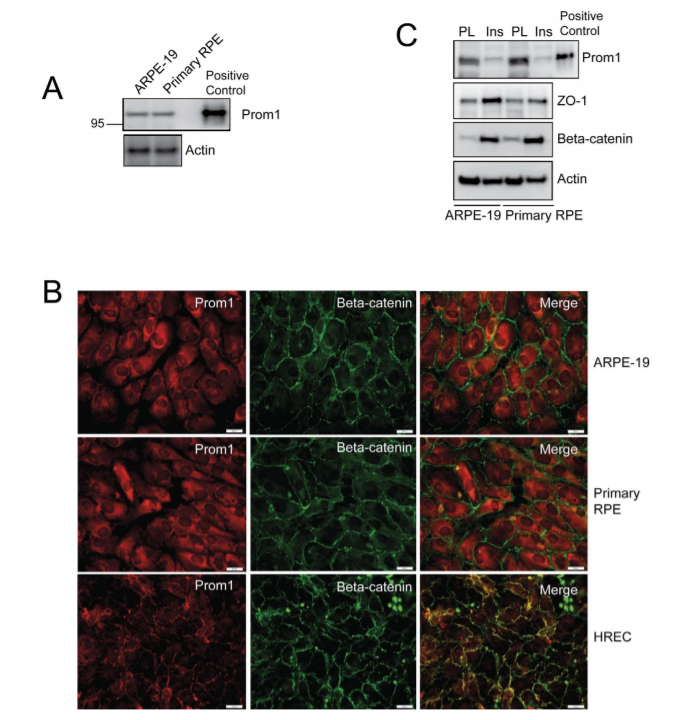
Figure 1. Expression and localization of Prom1 in human RPE.
Researchers investigated the expression and localization of Prom1 in both immortalized ARPE-19 cells and primary RPE cultures obtained from donor eyes. They observed similar levels of Prom1 expression were observed in ARPE-19 cells and primary RPE cultures. Immunofluorescence staining of Prom1 and β-catenin expression show that Prom1 is mainly distributed in the cytoplasm and perinuclear regions with some nuclear staining in the ARPE-19 and RPE cultures, which failed to co-localize with peripheral β-catenin. Differentiation of RPE was associated with the increased expression of ZO-1 and β-catenin and a decrease in Prom1 expression, compared to non-differentiated RPE, suggesting a correlation between reduced Prom1 expression and RPE differentiation.
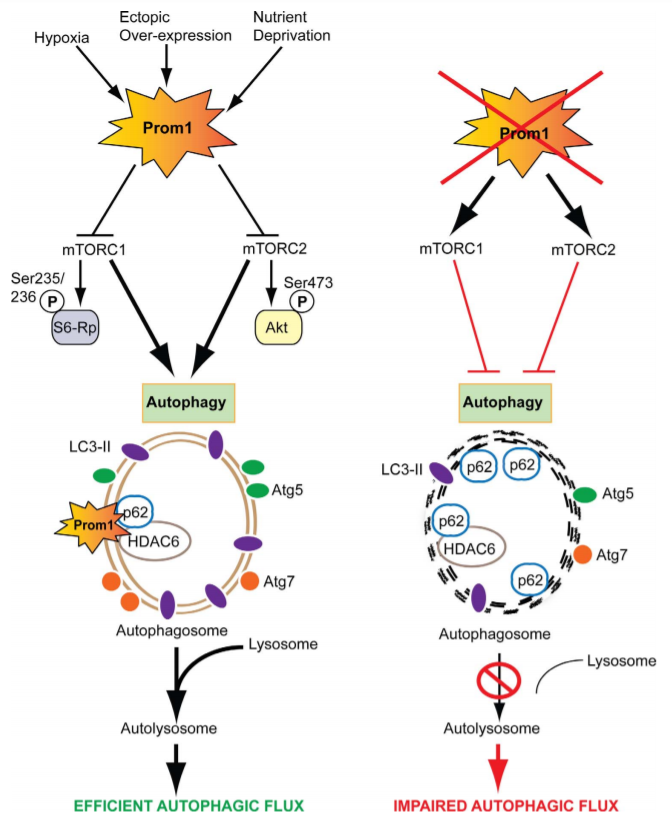
Figure 2: Schematic representation of molecular mechanisms regulating Prom1-dependent autophagy in the RPE
This study demonstrated that Prom1 is primarily a cytosolic protein in the RPE. Stress signals and physiological aging robustly increase autophagy with concomitant upregulation of Prom1 expression. Knockout of Prom1 increased mTORC1 and mTORC2 signaling, decreased autophagosome trafficking to the lysosome, increased p62 accumulation, and inhibited autophagic puncta induced by activators of autophagy. Conversely, ectopic overexpression of Prom1 inhibited mTORC1 and mTORC2 activities, and potentiated autophagy flux. Through interactions with p62 and HDAC6, Prom1 regulates autophagosome maturation and trafficking, suggesting a new cytoplasmic role of Prom1 in RPE function.
Study summary, Prom1 plays a key role in the regulation of autophagy via upstream suppression of mTOR signaling and also acting as a component of a macromolecular scaffold involving p62 and HDAC6.
A study of CRISPR-Cas9- and AON-based approaches to correct the splicing defect of the CLRN1 c.254-649T>G mutation
Usher syndrome (USH) is an autosomal recessive genetic disease characterized by deafness, vision loss due to retinitis pigmentosa (RP), and occasional vestibular dysfunction. In USH type 3 (USH3A), these symptoms occur in a progressive and variable manner. USH3A is commonly associated with mutations in CLRN1, a gene encoding the four-transmembrane-domain protein Clarin-1 of largely unknown function. CLRN1 mutations have an estimated prevalence of 1:100,000 individuals worldwide and recently a novel deep intronic CLRN1 founder mutation (c.254-649T>G) in Saudi Arabia. This splicing mutation generates an aberrant exon, which leads to a frameshift and a premature stop codon. A number of studies identifying deep intronic mutations in known or unknown genes increases, there is an unmet need for developing appropriate treatment strategies for this type of mutations.
In this study, researchers focused on the recently identified deep intronic c.254-649T>G CLRN1 splicing mutation and aimed to establish two causative treatment approaches: CRISPR-Cas9-mediated excision of the mutated intronic region and antisense oligonucleotide (AON)-mediated correction of mRNA splicing. The therapeutic potential of these approaches was validated in different cell types transiently or stably expressing CLRN1 minigenes. Both approaches led to substantial correction of the splice defect.
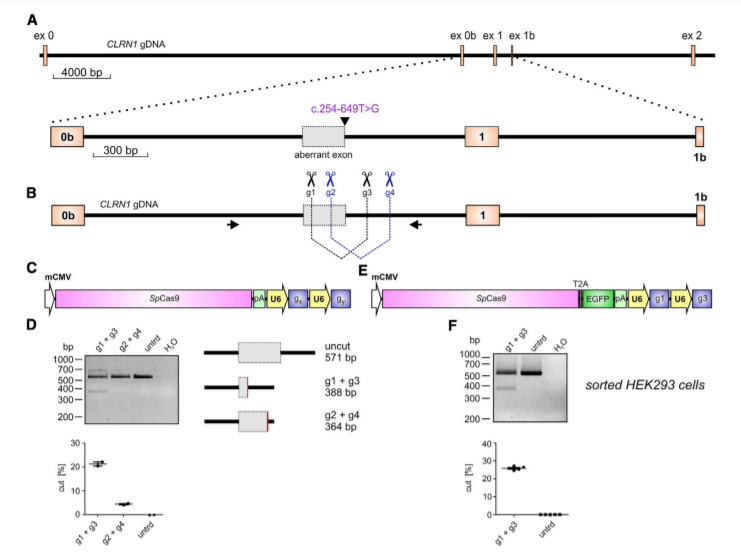
Figure 1. CRISPR-Cas9-Mediated CLRN1 Editing
The c.254-649T>G CLRN1 mutation is located in intron 0b and generates a novel splice donor site (SDS), which results in the inclusion of an aberrant exon in the mature mRNA (Fig.1A). To test the CRISPR-Cas9 efficiency for editing the CLRN1 locus, we designed four single guide RNAs (sgRNAs, g1–g4) targeting regions flanking the mutation (Fig1B). This CRISPR-Cas9 approach showed reasonable gene editing efficiency for the excision of the c.254-649T>G mutation from the native CLRN1 locus in HEK293 cells. The gene editing efficiency was around 25.9% ± 0.41%.
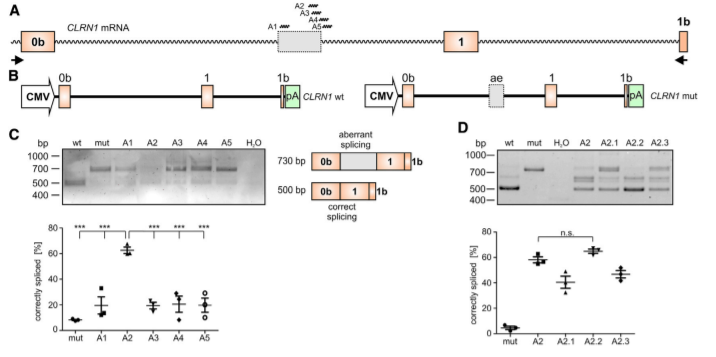
Figure 2. AON-Mediated Correction of CLRN1 mRNA Splicing
To test the potential of the antisense oligonucleotides (AONs) for correction of aberrant splicing caused by the c.254-649T>G mutation, the researcher designed five AONs (A1– A5) binding to different regions of the CLRN1 transcript. The use of AONs, particularly A2. (Fig2C, D), achieved a substantial correction of aberrant splicing caused by the c.254-649T>G mutation. AONs therefore another possible strategy, in addition to the CRISPR-Cas9 approach, for treating USH3A patients carrying this mutation.
It is well established that the efficiency of CRISPR-Cas9-mediated gene editing can be cell type-dependent. In this regard, targeting cells that express CLRN1 endogenously would be more therapeutically relevant. Therefore, the researcher tested two different retinal pigment epithelium (RPE)-derived cell lines for CLRN1 expression, i.e., ARPE-19, and human retinal pigment epithelial (hRPE) cells provided by the LMU Eye Hospital (Munich, Germany). The researcher found that the endogenous CLRN1 locus can be efficiently edited via CRISPR-Cas9, when delivered using a rAAV approach.
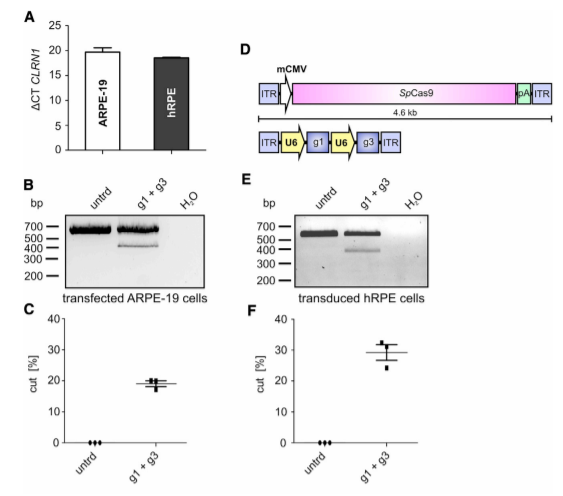
Figure 3. CRISPR-Cas9-Mediated CLRN1 Gene Editing in Transfected ARPE-19 and rAAV Transduced Human RPE Cells.
In summary, the therapeutic potential of CRISPR-Cas9 and AON-based strategies to correct the splicing of the CLRN1 c.254- 649T>G mutation, and offers an initial premise for further preclinical development, which could lead to the first clinical trials for USH3A patients.
Ubigene developed CRISPR-U™ which optimizes eukaryotic cells and animal gene-editing vectors and processes. The efficiency and accuracy are 10x higher than traditional methods. Contact us immediately to know about your research related services!

Reference:
Prominin-1 Is a Novel Regulator of Autophagy in the Human Retinal Pigment Epithelium. Invest Ophthalmol Vis Sci. Vol 58, 2017.
Antisense Oligonucleotide and CRISPRCas9-Mediated Rescue of mRNA Splicing for a Deep Intronic CLRN1 Mutation. Molecular Therapy: Nucleic Acids Vol. 21, 2020.





 Back To Top
Back To Top
Our Solution Specialists will contact you as soon as possible!