Scientist.com
Approved Supplier


 Gene Editing Services
Gene Editing Services
Ubigene has modified over 6000 genes from more than 300 cell lines with our exclusive innovation CRISPR-U™ technology. At the same time, we already provide customers with high quality gene-editing tools for cell or animal research worldwide.
 EZ-editor™
EZ-editor™With 15 years of experience, Ubigene has exclusively innovated and developed 6 product lines, fullfilling all kinds of needs from researchers. Experiment process simplified, efficiency improved, achieving our aim of 'Make genome editing easier'!
 Red cotton OmniCell bank
Red cotton OmniCell bank
It is a one-stop cell procurement plat-form for scientific researchers, gathering more than 5000 kinds of cell products in 5 categories, covering all fields and application directions of life science research.
 Red Cotton Gene knockout Project
Red Cotton Gene knockout Project
Ubigene exclusive KO Cell Line Bank, over 5000 KO cell lines, covering thousands of genes from 8 popular signaling pathways and nearly 100 diseases.
Location:Home > Application >
"Molecular Biologist Toolbox" E. coli with CRISPR form a new pace in biological studies
The microorganism Escherichia coli (E.coli) has a long history in the biotechnology industry and is still the microorganism of choice for most gene manipulation research. Factors such as its ability to grow fast using cheap media and availability of molecular tools to perform genetic manipulations are favorable for using E. coli as a model organism in molecular genetics. General people have known E. coli for the infectious nature of one particular strain (O157:H7) but few people are aware of how versatile and widely used it is in research as a common host for recombinant DNA (new genetic combinations from different species or sources). Several key discoveries in the field of molecular biology, including molecular genetics, were achieved using E. coli as a model organism. This includes an understanding of the genetic code, the mechanisms of DNA replication, the discovery of the genetic operon systems, and the creation of a genetically modified organism.
E. coli commonly found as a commensal in the human microbiota. However, the plasticity of its genome has led to the evolution of this organism into pathogenic strains able to cause diseases and syndromes of public health importance in humans and animals. Pathogenic E. coli are mainly divided into two groups depending on the disease location: extra-intestinal pathogenic E. coli (ExPEC) and intestinal pathogenic E. coli (InPEC). While ExPEC strains are associated mainly with neonatal meningitis (NMEC) and urinary tract infections (UPEC) in adults, InPEC strains, related to diarrheal disease, are subdivided into at least 6 well-known pathotypes: entero-pathogenic E. coli (EPEC), entero-hemorrhagic E. coli (EHEC), entero-invasive E. coli (EIEC), entero-aggregative E. coli (EAEC), diffusely adherent E. coli (DAEC) and entero-toxigenic E. coli (ETEC). E. coli virulence factors, mechanisms of infection, interaction with the enterocyte, tissue tropism, vaccine development, etc. are the key research area of gut microbiota.
Of all the model organisms and tools for genetic modification available, Escherichia coli stands out as a model organism and what we would like to call the “molecular biologist toolbox”. Regarding genetic modifications and tools for modifying E. coli is highly potential to generate plasmid vectors, single and multiple gene knockouts, whole-genome editing, biosensor (generation and applications), synthetic gene circuits, and genomes.
1. Biosensor: Genome engineering, plasmid or chromosomal constructs are useful for the detection of environmental traits, hazards, measuring cellular processes as any standard reporter system. Biosensors for detecting different traits like oxidants, DNA damaging compounds, membrane-damaging compounds, protein-damaging compounds, aromatic compounds, xenobiotics, antibiotics, etc.
2. Synthetic biology & metabolic engineering: Development of designed microorganism, the production of novel compounds from existing gene clusters (through module shuffling, mutagenesis, or deletions), or by the introduction of novel environmentally sequenced gene clusters for heterologous production, generate a flexible platform for the efficient production of molecules ( for the pharmaceutical industry, food industry, and biofuel industry, etc.)
3. Vaccine developments: Study on virulence, virome tracking, generate mutant virus, promising antigens identification, bacterial vaccination (YncE, expressed by different E. coli strains, including ETEC, EHEC, EAEC, and UPEC pathotypes).
CRISPR-B™ for efficient gene editing in E. coli
Genome editing and manipulation often revolutionize the understanding, exploitation, and control of microbial species. E. coli represents a model organism for the biological study of biofilm formation, drug target, generate mutant virus, virulence study, vaccine development, virome tracking, biosensors for detecting different traits (aromatic compounds, xenobiotics, antibiotic, DNA damaging compounds, etc.), and metabolic engineering for the production of metabolites and molecules (pharmaceutical industry, food industry, and biofuel industry, etc.).
However, a traditional method for E. coli genetic manipulation required multi-steps selection process to generate mutant and leave a scar sequence in the place of deleted gene. So, the traditional genetic manipulation methods for engineering E. coli, are still time consuming and laborious. Ubigene developed CRISPR-B™ for gene manipulation of E. coli. Thereby, possible to achieve a one-step seamless genome editing in E. coli species with the utilization of the CRISPR/Cas9 system. The CRISPR/Cas9 and the λ-Red recombination systems used for rapid, precise, and seamless genetic manipulation in E. coli. Ubigene can customize the gene-editing in E. coli as well as can generate various genes modification in microbes.
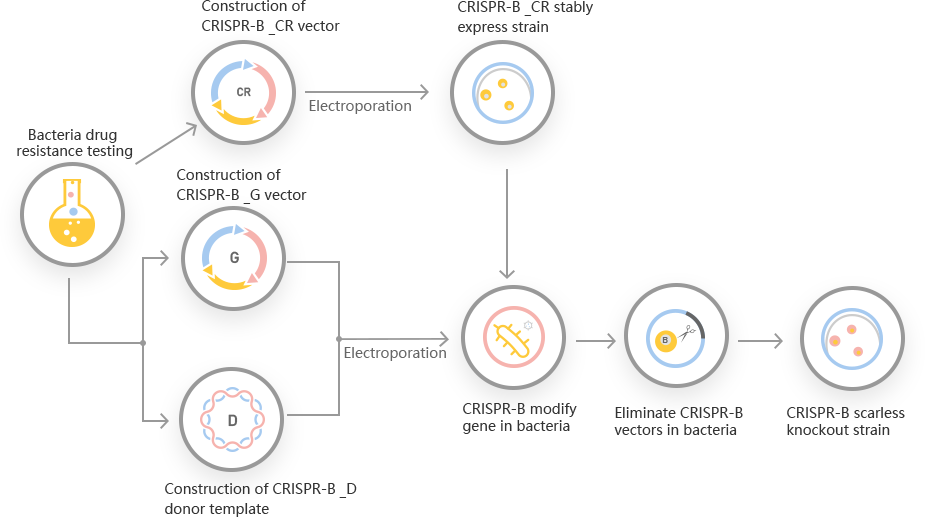
CRISPR-B™ workflow for E. coli genetic engineering
n‑butanol production from xylose by CRISPR/Cas9 mediated engineered Escherichia coli
Biofuels produced from lignocellulosic biomass has been considered a feasible alternative to fossil fuels. Biobutanol is a renewable alternative that possesses various important advantages compared to ethanol, including higher energy content, hydrophobicity, octane number, and lower vapor pressure, which provide better blending ability with gasoline at any fraction. Besides, it can be used directly in conventional engines without any modification. In this study, the researcher used CRISPR/Cas9 tool to integrate a large portion of DNA fragment carrying a butanol pathway into E. coli genome an efficient xylose-utilizing host, i.e., E. coli SSK42 (Fig. 1), and produce butanol from xylose as a sole carbon source in a defined medium. They considered the Clostridial-based fermentative pathway for n-butanol production in E. coli, except for the first pathway enzyme acetyl-CoA acetyltransferase (THL) from E. coli itself and the intermediate enzyme trans-enoyl coenzyme A (CoA) reductase (TER) from the T. denticola (Fig. 1).
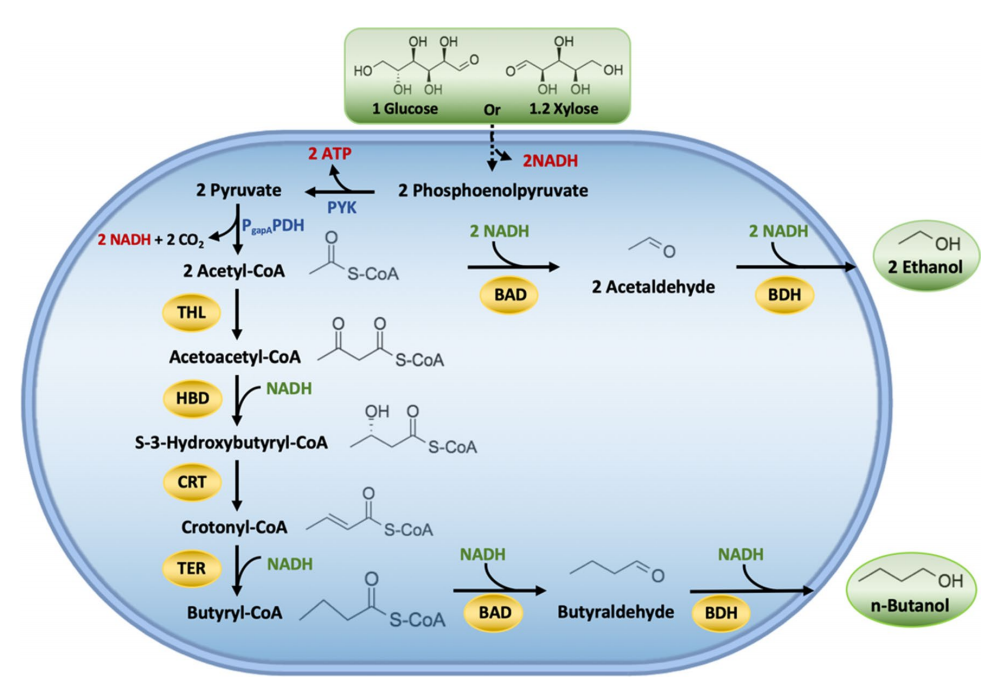
Fig.1 Schematic representation of butanol production pathway in engineered E. coli ASA02 strain.
The researcher optimized a synthetic construct that contains two operons under the control of T5 promoter. The first operon contains atoB, hbd and crt genes, and the second operon contains adhE2 and ter genes. A homology arm of 600 bp of an intergenic region SS9 (safe site 9) was introduced to the 5′ and 3′ ends of the construct for genomic integration via CRISPR/Cas9 technique (Fig 2).
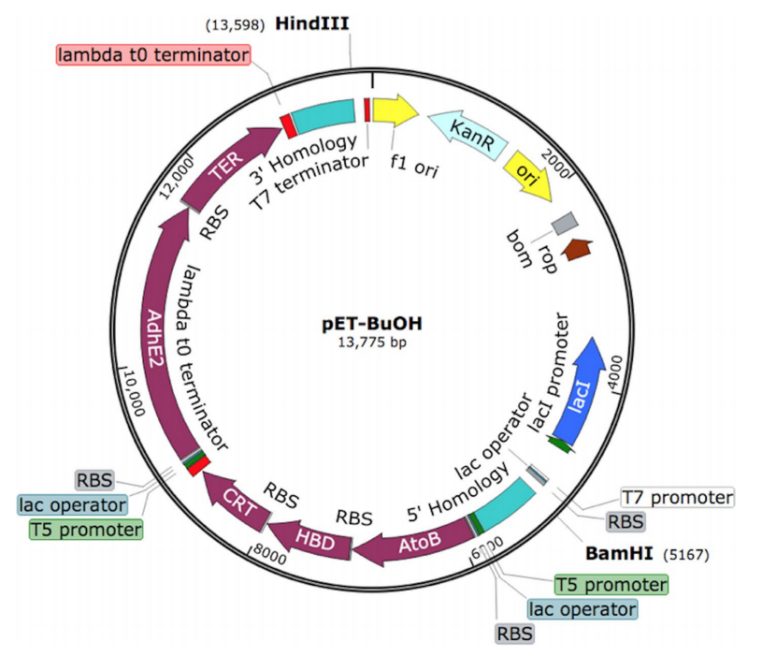
Fig 2: The optimized synthetic construct of an essential set of genes for butanol production.
For producing butanol from xylose, they considered a xylose-fermenting ethanologenic E. coli strain SSK42 as a platform host for metabolic engineering. They deleted the endogenous adhE gene of SSK42 via P1 phage transduction and then integrated synthetic butanol pathway cassette in its genome using CRISPR/Cas9 technique to divert carbon flux towards butanol (Fig. 1). The newly engineered strain ASA02 was evaluated for its ability to produce butanol via. induction of butanol operons using 0.01 mM IPTG and optimal xylose concentration for butanol production. (Fig. 3a, b).
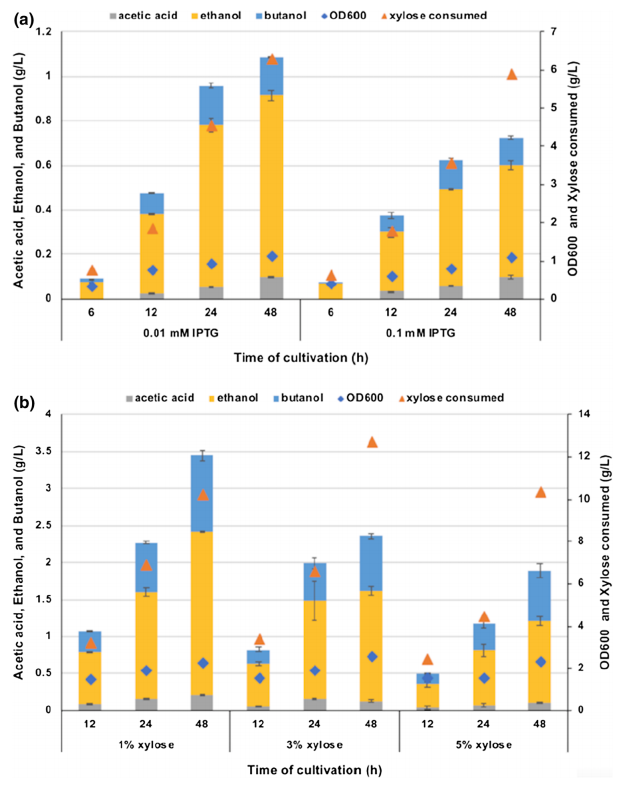
Fig 3: Comparison of the effect of different IPTG and xylose concentrations on the production of alcohol in E. coli ASA02 strain.
Different fermentation strategies were performed to produce butanol from xylose sugar in a defined AM1 medium. Cells were grown in AM1 medium with xylose under the microaerobic condition in a bioreactor (Fig4a). Bacterial cells were grown in AM1 medium with xylose in a bioreactor under the aerobic condition for 19 h then shifted to anaerobic condition (Fig 4b).
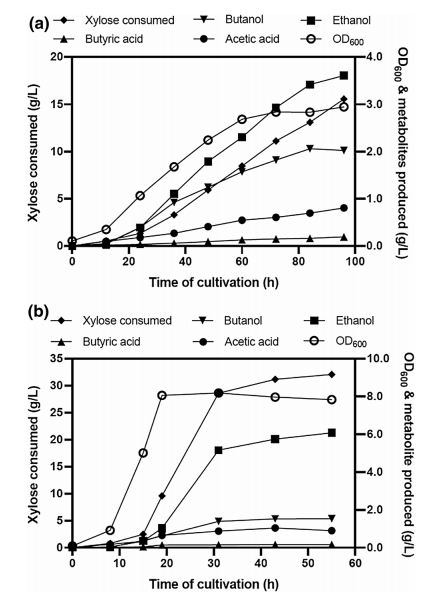
Fig. 4 Batch fermentation profile of E. coli ASA02 strain.
They performed a fed-batch fermentation where initiated batch fermentation in 1% xylose and then further added xylose intermittently to maintain the level ~0.5–1% in the bioreactor. They tested the impact of airflow rate on butanol production under the microaerobic condition and this experiment was conducted by supplying airflow in the headspace at the rate ranging from 0.03 to 1.66 VVM. They observe that the butanol production increased when the airflow rate was increased from 0.03 to 0.08 VVM and then decreased upon a subsequent increase in the airflow rate. They found that a consistent increase in butanol formation throughout the fermentation at 0.08 VVM of airflow rate, which reached 4.3 g/L at 188 h with 0.13 g/g xylose yield (Fig 5).
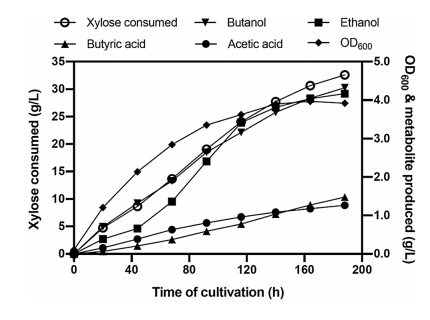
Fig 5: Fed-batch fermentation profile of E. coli ASA02 strain. Cells were grown in AM1 medium with xylose under micro-aerobic conditions.
CRISPR/Cas9 mediated Reversal of mcr-1-Mediated Colistin Resistance in Escherichia coli
Antibiotics have significant impacts on health care since their first introduction in the early 20th century. Antimicrobial resistance genes (ARGs) often present on mobile genetic elements, can be transferred among bacteria, which led to the global prevalence of antibiotic-resistant bacteria. The plasmid-mediated mobilized colistin resistance gene (mcr-1) was discovered in 2015. Horizontal transfer of mcr-1 gene to diverse bacterial species poses a serious threat to public health, which urgently needs the introduction of novel antimicrobial strategies. Herein, the researcher used CRISPR/Cas9 system to sensitize bacteria to colistin and reduce the propagation of the mcr-1 gene by curing mcr-1-harboring plasmid in Escherichia coli (E. coli). Re-sensitize colistin-resistant E. coli by curing mcr-1-carrying plasmid is considered as a potentially effective method to resist the dissemination of colistin resistance genes.
To achieve re-sensitize of E. coli, they constructed the high copy number plasmid pUC19-mcr-1 and recombinant plasmid pCas9-m1 or pCas9-m2, which contain 20 nt or 30 nt sgRNA sequences targeted to mcr-1, respectively (Fig 1).
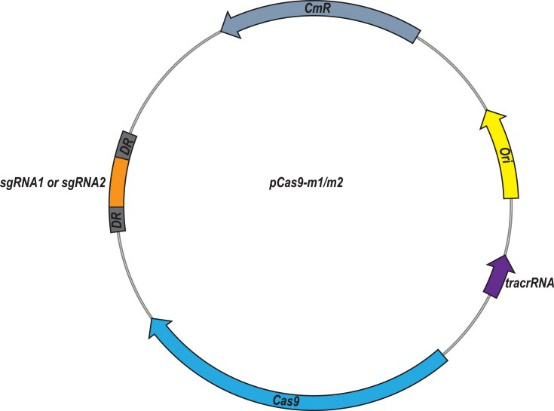
Fig 1: Plasmid map of pCas9-m1/m2 targeted to mcr-1 gene. The pCas9-m1/m2 was constructed by inserting a spacer targeting the mcr-1 gene.
CRISPR/Cas9 system can efficiently eliminate plasmid from E. coli, the control vector pCas9, and the engineered vector pCas9-m1 were transformed into competent E. coli C600+pHNSHP45. The results showed that the 582bp fragment indicating the presence of mcr-1 gene failed to be amplified from 18/24 colonies transformed with pCas9-m1(Fig. 2A). The competent cells of E. coli C600+pUC19-mcr-1 were used as host and transformed with pCas9-m1. The colony PCR showed that 24 colonies were mcr-1 negative (Fig. 2B). These results indicate that mcr-1-harboring plasmids could be efficiently eliminated.
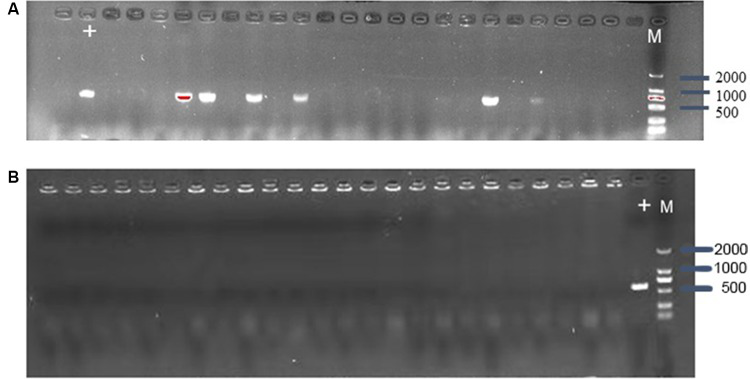
Fig-2: (A) Confirmation of mcr-1 gene presence in E. coli C600+pHNSHP45 by PCR amplification with primer mcr-1-JD-F/R. (M- marker, the + lane is strain transformed with pCas9 as a positive control. The other lanes strain transformed with pCas9-m1). (B) Confirmation of mcr-1 gene elimination in C600+pUC19-mcr-1 by PCR amplification with primer mcr-1-JD-F/R.
To further evaluate editing efficiency, the relative copy number of plasmids at each time point in pCas9-m1 or pCas9-m2 treated group and pCas9 control group was determined by qPCR. Significant differences in plasmid copy numbers can be seen between the experimental and control groups (Fig. 3). These results clearly indicate that the engineered CRISPR/Cas9 system can effectively eliminate mcr-1 gene and the length of sgRNA sequence has no significant effect.
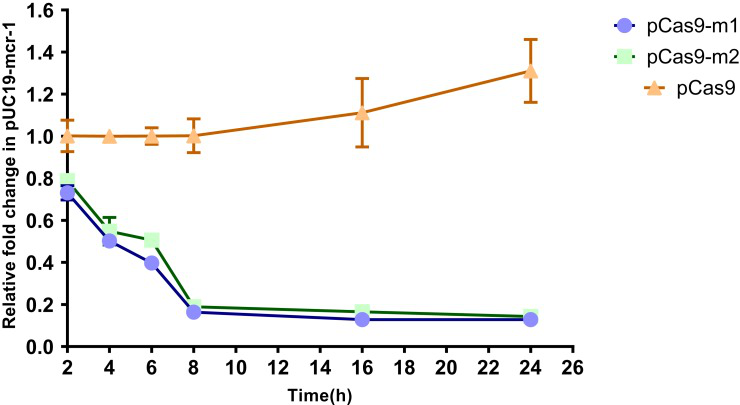
Fig-3. The relative copy number of plasmids pUC19-mcr-1 at each time point. pCas9-m1 and pCas9-m2 transformed into competent E. coli C600+pUC19-mcr-1 serve as experimental groups.
Bacterial conjugation is an important way to mediate the horizontal transfer of resistance genes. The CRISPR-Cas system is an adaptive immune system of bacteria and archaea, which can generate immune memory to foreign DNA. The engineered CRISPR/Cas9 system in bacteria could block plasmid conjugation. The researcher found that the engineered CRISPR/Cas9 system in the recipient cell could block the horizontal transfer of the conjugative plasmid pHNSHP45 (Fig. 4).
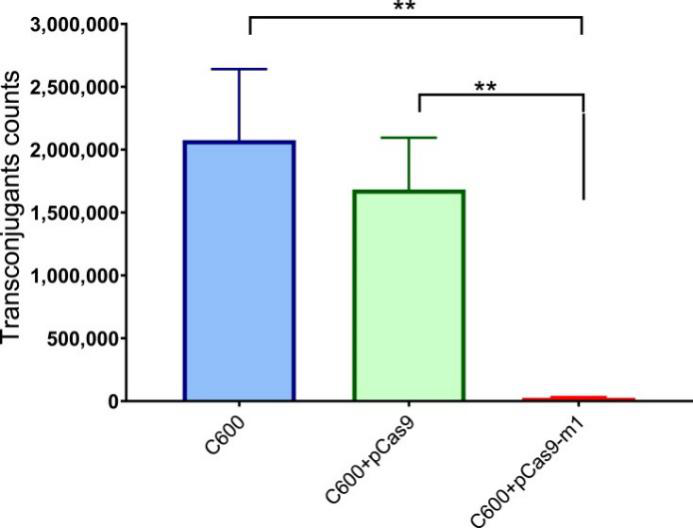
Fig. 4. E. coli strain C600 harboring pCas9-m-1 limited the conjugation of pHNSHP45. Colistin-resistant E. coli C600+ pHNSHP45 as donor and E. coli C600, chloramphenicol-resistant, E. coli C600+pCas9-m1 or C600+pCas9 as the recipient strain.
when pCas9-m1 was transformed into E. coli C600+pHNSHP45, there were 6/24 colonies still resistant to colistin. To explore the basis of escape, they extracted plasmids from escape mutants and sequencing. The found that mutants escaped through spacer mutations in the CRISPR locus, deletions in tracrRNA, and transposon insertions in Cas9 as presented in fig.5.
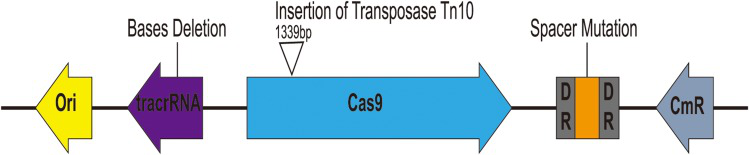
Fig. 5 Characterization of escape mutants that tolerated transformation of pCas9-m1 construct.
In summary, an engineered CRISPR/Cas9 system could be used to efficiently re-sensitize resistant isolates to colistin and reduce the horizontal transfer of the conjugative plasmid pHNSHP45. Together, the CRISPR/Cas9 system is an attractive alternative against antibiotic resistance, and highly potential for clinical application.
Ubigene developed CRISPR-U™ which optimizes eukaryotic cells and animal gene-editing vectors and processes. The efficiency and accuracy are 10x higher than traditional methods. Contact us immediately to know about your research related services!

Case study 1 Reference:
CRISPR/Cas9‑mediated engineering of Escherichia coli for n‑butanol production from xylose in defined medium. J Ind Microbiol Biotechnol. 2019;46 (7):965-975.
Reversal of mcr-1-Mediated Colistin Resistance in Escherichia coli by CRISPR/Cas9 System. Infect Drug Resist. 2020; 13: 1171–1178.
"Molecular Biologist Toolbox" E. coli with CRISPR form a new pace in biological studies
The microorganism Escherichia coli (E.coli) has a long history in the biotechnology industry and is still the microorganism of choice for most gene manipulation research. Factors such as its ability to grow fast using cheap media and availability of molecular tools to perform genetic manipulations are favorable for using E. coli as a model organism in molecular genetics. General people have known E. coli for the infectious nature of one particular strain (O157:H7) but few people are aware of how versatile and widely used it is in research as a common host for recombinant DNA (new genetic combinations from different species or sources). Several key discoveries in the field of molecular biology, including molecular genetics, were achieved using E. coli as a model organism. This includes an understanding of the genetic code, the mechanisms of DNA replication, the discovery of the genetic operon systems, and the creation of a genetically modified organism.
E. coli commonly found as a commensal in the human microbiota. However, the plasticity of its genome has led to the evolution of this organism into pathogenic strains able to cause diseases and syndromes of public health importance in humans and animals. Pathogenic E. coli are mainly divided into two groups depending on the disease location: extra-intestinal pathogenic E. coli (ExPEC) and intestinal pathogenic E. coli (InPEC). While ExPEC strains are associated mainly with neonatal meningitis (NMEC) and urinary tract infections (UPEC) in adults, InPEC strains, related to diarrheal disease, are subdivided into at least 6 well-known pathotypes: entero-pathogenic E. coli (EPEC), entero-hemorrhagic E. coli (EHEC), entero-invasive E. coli (EIEC), entero-aggregative E. coli (EAEC), diffusely adherent E. coli (DAEC) and entero-toxigenic E. coli (ETEC). E. coli virulence factors, mechanisms of infection, interaction with the enterocyte, tissue tropism, vaccine development, etc. are the key research area of gut microbiota.
Of all the model organisms and tools for genetic modification available, Escherichia coli stands out as a model organism and what we would like to call the “molecular biologist toolbox”. Regarding genetic modifications and tools for modifying E. coli is highly potential to generate plasmid vectors, single and multiple gene knockouts, whole-genome editing, biosensor (generation and applications), synthetic gene circuits, and genomes.
1. Biosensor: Genome engineering, plasmid or chromosomal constructs are useful for the detection of environmental traits, hazards, measuring cellular processes as any standard reporter system. Biosensors for detecting different traits like oxidants, DNA damaging compounds, membrane-damaging compounds, protein-damaging compounds, aromatic compounds, xenobiotics, antibiotics, etc.
2. Synthetic biology & metabolic engineering: Development of designed microorganism, the production of novel compounds from existing gene clusters (through module shuffling, mutagenesis, or deletions), or by the introduction of novel environmentally sequenced gene clusters for heterologous production, generate a flexible platform for the efficient production of molecules ( for the pharmaceutical industry, food industry, and biofuel industry, etc.)
3. Vaccine developments: Study on virulence, virome tracking, generate mutant virus, promising antigens identification, bacterial vaccination (YncE, expressed by different E. coli strains, including ETEC, EHEC, EAEC, and UPEC pathotypes).
CRISPR-B™ for efficient gene editing in E. coli
Genome editing and manipulation often revolutionize the understanding, exploitation, and control of microbial species. E. coli represents a model organism for the biological study of biofilm formation, drug target, generate mutant virus, virulence study, vaccine development, virome tracking, biosensors for detecting different traits (aromatic compounds, xenobiotics, antibiotic, DNA damaging compounds, etc.), and metabolic engineering for the production of metabolites and molecules (pharmaceutical industry, food industry, and biofuel industry, etc.).
However, a traditional method for E. coli genetic manipulation required multi-steps selection process to generate mutant and leave a scar sequence in the place of deleted gene. So, the traditional genetic manipulation methods for engineering E. coli, are still time consuming and laborious. Ubigene developed CRISPR-B™ for gene manipulation of E. coli. Thereby, possible to achieve a one-step seamless genome editing in E. coli species with the utilization of the CRISPR/Cas9 system. The CRISPR/Cas9 and the λ-Red recombination systems used for rapid, precise, and seamless genetic manipulation in E. coli. Ubigene can customize the gene-editing in E. coli as well as can generate various genes modification in microbes.
CRISPR-B™ workflow for E. coli genetic engineering
n‑butanol production from xylose by CRISPR/Cas9 mediated engineered Escherichia coli
Biofuels produced from lignocellulosic biomass has been considered a feasible alternative to fossil fuels. Biobutanol is a renewable alternative that possesses various important advantages compared to ethanol, including higher energy content, hydrophobicity, octane number, and lower vapor pressure, which provide better blending ability with gasoline at any fraction. Besides, it can be used directly in conventional engines without any modification. In this study, the researcher used CRISPR/Cas9 tool to integrate a large portion of DNA fragment carrying a butanol pathway into E. coli genome an efficient xylose-utilizing host, i.e., E. coli SSK42 (Fig. 1), and produce butanol from xylose as a sole carbon source in a defined medium. They considered the Clostridial-based fermentative pathway for n-butanol production in E. coli, except for the first pathway enzyme acetyl-CoA acetyltransferase (THL) from E. coli itself and the intermediate enzyme trans-enoyl coenzyme A (CoA) reductase (TER) from the T. denticola (Fig. 1).
Fig.1 Schematic representation of butanol production pathway in engineered E. coli ASA02 strain.
The researcher optimized a synthetic construct that contains two operons under the control of T5 promoter. The first operon contains atoB, hbd and crt genes, and the second operon contains adhE2 and ter genes. A homology arm of 600 bp of an intergenic region SS9 (safe site 9) was introduced to the 5′ and 3′ ends of the construct for genomic integration via CRISPR/Cas9 technique (Fig 2).
Fig 2: The optimized synthetic construct of an essential set of genes for butanol production.
For producing butanol from xylose, they considered a xylose-fermenting ethanologenic E. coli strain SSK42 as a platform host for metabolic engineering. They deleted the endogenous adhE gene of SSK42 via P1 phage transduction and then integrated synthetic butanol pathway cassette in its genome using CRISPR/Cas9 technique to divert carbon flux towards butanol (Fig. 1). The newly engineered strain ASA02 was evaluated for its ability to produce butanol via. induction of butanol operons using 0.01 mM IPTG and optimal xylose concentration for butanol production. (Fig. 3a, b).
Fig 3: Comparison of the effect of different IPTG and xylose concentrations on the production of alcohol in E. coli ASA02 strain.
Different fermentation strategies were performed to produce butanol from xylose sugar in a defined AM1 medium. Cells were grown in AM1 medium with xylose under the microaerobic condition in a bioreactor (Fig4a). Bacterial cells were grown in AM1 medium with xylose in a bioreactor under the aerobic condition for 19 h then shifted to anaerobic condition (Fig 4b).
Fig. 4 Batch fermentation profile of E. coli ASA02 strain.
They performed a fed-batch fermentation where initiated batch fermentation in 1% xylose and then further added xylose intermittently to maintain the level ~0.5–1% in the bioreactor. They tested the impact of airflow rate on butanol production under the microaerobic condition and this experiment was conducted by supplying airflow in the headspace at the rate ranging from 0.03 to 1.66 VVM. They observe that the butanol production increased when the airflow rate was increased from 0.03 to 0.08 VVM and then decreased upon a subsequent increase in the airflow rate. They found that a consistent increase in butanol formation throughout the fermentation at 0.08 VVM of airflow rate, which reached 4.3 g/L at 188 h with 0.13 g/g xylose yield (Fig 5).
Fig 5: Fed-batch fermentation profile of E. coli ASA02 strain. Cells were grown in AM1 medium with xylose under micro-aerobic conditions.
CRISPR/Cas9 mediated Reversal of mcr-1-Mediated Colistin Resistance in Escherichia coli
Antibiotics have significant impacts on health care since their first introduction in the early 20th century. Antimicrobial resistance genes (ARGs) often present on mobile genetic elements, can be transferred among bacteria, which led to the global prevalence of antibiotic-resistant bacteria. The plasmid-mediated mobilized colistin resistance gene (mcr-1) was discovered in 2015. Horizontal transfer of mcr-1 gene to diverse bacterial species poses a serious threat to public health, which urgently needs the introduction of novel antimicrobial strategies. Herein, the researcher used CRISPR/Cas9 system to sensitize bacteria to colistin and reduce the propagation of the mcr-1 gene by curing mcr-1-harboring plasmid in Escherichia coli (E. coli). Re-sensitize colistin-resistant E. coli by curing mcr-1-carrying plasmid is considered as a potentially effective method to resist the dissemination of colistin resistance genes.
To achieve re-sensitize of E. coli, they constructed the high copy number plasmid pUC19-mcr-1 and recombinant plasmid pCas9-m1 or pCas9-m2, which contain 20 nt or 30 nt sgRNA sequences targeted to mcr-1, respectively (Fig 1).
Fig 1: Plasmid map of pCas9-m1/m2 targeted to mcr-1 gene. The pCas9-m1/m2 was constructed by inserting a spacer targeting the mcr-1 gene.
CRISPR/Cas9 system can efficiently eliminate plasmid from E. coli, the control vector pCas9, and the engineered vector pCas9-m1 were transformed into competent E. coli C600+pHNSHP45. The results showed that the 582bp fragment indicating the presence of mcr-1 gene failed to be amplified from 18/24 colonies transformed with pCas9-m1(Fig. 2A). The competent cells of E. coli C600+pUC19-mcr-1 were used as host and transformed with pCas9-m1. The colony PCR showed that 24 colonies were mcr-1 negative (Fig. 2B). These results indicate that mcr-1-harboring plasmids could be efficiently eliminated.
Fig-2: (A) Confirmation of mcr-1 gene presence in E. coli C600+pHNSHP45 by PCR amplification with primer mcr-1-JD-F/R. (M- marker, the + lane is strain transformed with pCas9 as a positive control. The other lanes strain transformed with pCas9-m1). (B) Confirmation of mcr-1 gene elimination in C600+pUC19-mcr-1 by PCR amplification with primer mcr-1-JD-F/R.
To further evaluate editing efficiency, the relative copy number of plasmids at each time point in pCas9-m1 or pCas9-m2 treated group and pCas9 control group was determined by qPCR. Significant differences in plasmid copy numbers can be seen between the experimental and control groups (Fig. 3). These results clearly indicate that the engineered CRISPR/Cas9 system can effectively eliminate mcr-1 gene and the length of sgRNA sequence has no significant effect.
Fig-3. The relative copy number of plasmids pUC19-mcr-1 at each time point. pCas9-m1 and pCas9-m2 transformed into competent E. coli C600+pUC19-mcr-1 serve as experimental groups.
Bacterial conjugation is an important way to mediate the horizontal transfer of resistance genes. The CRISPR-Cas system is an adaptive immune system of bacteria and archaea, which can generate immune memory to foreign DNA. The engineered CRISPR/Cas9 system in bacteria could block plasmid conjugation. The researcher found that the engineered CRISPR/Cas9 system in the recipient cell could block the horizontal transfer of the conjugative plasmid pHNSHP45 (Fig. 4).
Fig. 4. E. coli strain C600 harboring pCas9-m-1 limited the conjugation of pHNSHP45. Colistin-resistant E. coli C600+ pHNSHP45 as donor and E. coli C600, chloramphenicol-resistant, E. coli C600+pCas9-m1 or C600+pCas9 as the recipient strain.
when pCas9-m1 was transformed into E. coli C600+pHNSHP45, there were 6/24 colonies still resistant to colistin. To explore the basis of escape, they extracted plasmids from escape mutants and sequencing. The found that mutants escaped through spacer mutations in the CRISPR locus, deletions in tracrRNA, and transposon insertions in Cas9 as presented in fig.5.
Fig. 5 Characterization of escape mutants that tolerated transformation of pCas9-m1 construct.
In summary, an engineered CRISPR/Cas9 system could be used to efficiently re-sensitize resistant isolates to colistin and reduce the horizontal transfer of the conjugative plasmid pHNSHP45. Together, the CRISPR/Cas9 system is an attractive alternative against antibiotic resistance, and highly potential for clinical application.
Ubigene developed CRISPR-U™ which optimizes eukaryotic cells and animal gene-editing vectors and processes. The efficiency and accuracy are 10x higher than traditional methods. Contact us immediately to know about your research related services!
Case study 1 Reference:
CRISPR/Cas9‑mediated engineering of Escherichia coli for n‑butanol production from xylose in defined medium. J Ind Microbiol Biotechnol. 2019;46 (7):965-975.
Reversal of mcr-1-Mediated Colistin Resistance in Escherichia coli by CRISPR/Cas9 System. Infect Drug Resist. 2020; 13: 1171–1178.









