IF=26.8 | Ubigene's KO Cells Help Discover New Targets for Steatotic Liver Disease

IF=26.8 | Ubigene's KO Cells Help Discover New Targets for Steatotic Liver Disease

Background
Increasing evidence suggests that innate immunity, especially macrophage activation, significantly promotes the development of metabolic dysfunction-associated steatotic liver disease (MASLD). Therefore, macrophage involvement in inflammation and interaction with hepatocytes and hematopoietic stem cells are potential therapeutic targets for MASLD. MicroRNAs (miRNAs) are a class of small non-coding RNAs that can bind to and degrade target mRNA. Abnormal miRNA expression regulates downstream genes crucial for the occurrence or progression of MASLD. In addition to post-transcriptional regulation of target genes, there is evidence that miRNAs exist in the nucleus of cells and play a regulatory role. Previous literature has reported that nuclear miR-204-3p in macrophages inhibits CD36 transcription and the formation of foam cell derived from macrophages, highlighting the nuclear function of miR-204-3p in regulating gene transcription, which warrants further investigation. Furthermore, miR-204-3p has anti-inflammatory effects in several inflammation-related disease models, but its significance in MASLD remains unclear.
Abstract
Recently, the team led by Sijia Liang from Sun Yat-sen University published a research paper titled “Nuclear miR-204-3p mitigates metabolic dysfunction-associated steatotic liver disease in mice” in Journal of Hepatology. This study revealed that miR-204-3p inhibits macrophage inflammation, coordinates macrophage interactions with hepatocytes and HSCs, and improves fatty liver disease. Macrophage miR-204-3p may be a therapeutic target for MASLD. In this study, TLR4 gene knockout THP-1 cells constructed by Ubigene were used to verify whether the anti-inflammatory effects of miR-204-3p depend on the inhibition of the TLR4/JNK signaling pathway.
Results & Discussion
Quantitative reverse transcription PCR shows that miR-204-3p expression decreased in the liver and macrophages of high-fat diet (HFD) mice and ob/ob mice. Small RNA sequencing analysis of samples obtained from participants shows that miR-204-3p levels were also downregulated in liver tissue and peripheral blood mononuclear cells (PBMCs) of MASLD patients, and miR-204-3p levels in PBMCs were negatively correlated with the severity of liver disease and damage. In summary, these results suggest that macrophage miR-204-3p levels are associated with hepatic steatosis, inflammation, liver injury, and disease occurrence.

Fig. 1. Reduced miR-204-3p levels in MASLD samples correlate with disease severity.
Considering the significant decrease in macrophage miR-204-3p levels during the progression of hepatic steatosis, the potential impact on fatty liver disease was studied. By injecting adeno-associated viruses overexpressing miR-204-3p (AAV-204-3p) into HFD mice, it was found that overexpression of miR-204-3p in macrophages alleviated hepatic steatosis, inflammation, and insulin resistance in mice, improving liver-related indicators. Similar results were obtained in methionine-choline-deficient (MCD) diet-induced MASH mouse models. Bone marrow transplantation experiments further confirmed the negative regulatory role of macrophage miR-204-3p in fatty liver disease. Overall, macrophage miR-204-3p improves experimental fatty liver disease in mice.

Fig. 2. Macrophage miR-204-3p ameliorates experimental steatohepatitis in mice.
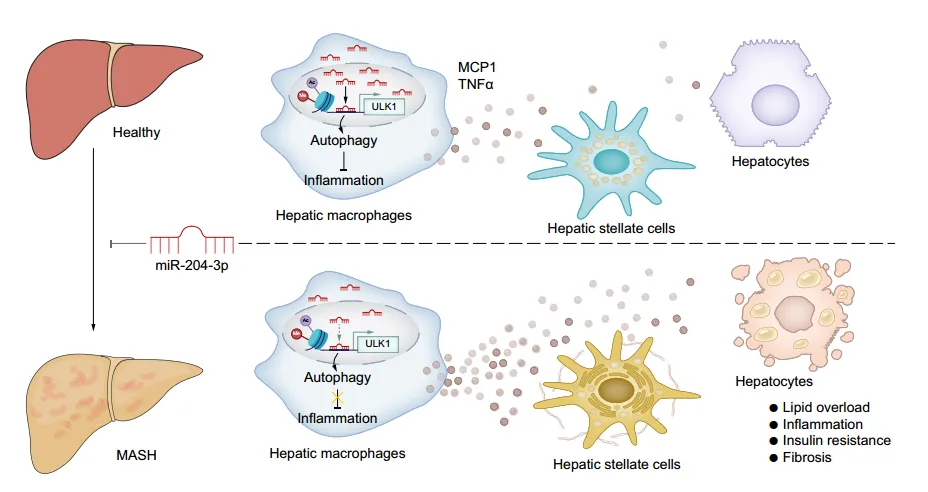
The authors established a co-culture system of hepatic macrophages with primary hepatocytes or hematopoietic stem cells. Co-culture experiments showed that co-culturing with macrophages from HFD mice increased lipid deposition, inflammation, and insulin resistance in hepatocytes, and increased fibrotic gene expression in HSCs; whereas co-culturing with macrophages overexpressing miR-204-3p attenuated these changes. Knockdown of miR-204-3p enhanced JNK phosphorylation and secretion of MCP1 and TNFα in macrophages, but not in Tlr4-/-THP-1 cells, indicating that TLR4/JNK signaling inhibition is necessary for the anti-inflammatory effects of miR-204-3p. In macrophages overexpressing miR-204-3p, TLR4 expression and JNK phosphorylation were decreased, and secretion of inflammatory factors was reduced, thereby inhibiting hepatocyte steatosis and HSC activation. These results indicate that macrophage miR-204-3p reduces the secretion of pro-inflammatory cytokines, inhibiting hepatocyte lipid accumulation and HSC fibrotic activation.

Fig. 3. Macrophage miR-204-3p attenuates hepatocyte lipid accumulation and inflammation.
To elucidate the molecular mechanism of the anti-inflammatory effect of miR-204-3p in macrophages, the authors performed RNA sequencing on macrophages with upregulated miR-204-3p. They found that differentially expressed genes enriched in epigenetic modification-related gene ontology terms, with downregulated genes highly related to MASLD pathways. Fluorescence in situ hybridization revealed significant nuclear localization of miR-204-3p in liver macrophages, with reduced nuclear miR-204-3p levels in the liver of MASH and HFD-fed mice. The authors identified metabolism-related miR-204-3p regulatory genes and potential target genes ULK1 and NFXL1, and confirmed that ULK1 expression continuously increases after miR-204-3p overexpression. RNA hybridization prediction showed that miR-204-3p upregulates ULK1 promoter activity and transcript levels by binding to a conserved site at -153bp of the ULK1 transcription start site. Additionally, miR-204-3p promotes ULK1 promoter enrichment of RNAPII and H3K27 acetylation and H3K4 trimethylation in HMDMs, thereby enhancing ULK1 transcription.

Fig. 4. miR-204-3p acts in the nucleus and activates ULK1 transcription.
Given the important role of ULK1 in the initiation of autophagy, the authors investigated whether miR-204-3p restricts macrophage inflammation in an autophagy-dependent manner. They found that inhibiting autophagy reversed the suppression of the inflammatory response by miR-204-3p, while inducing autophagy enhanced the inactivation of the TLR4/JNK signaling pathway, indicating that miR-204-3p is associated with autophagy. Through experiments, the authors confirmed that miR-204-3p promotes autophagosome formation and fusion with lysosomes, increasing autophagic flux. Additionally, overexpression of miR-204-3p upregulated the expression of 15 autophagy-related genes, with a significant increase in the mRNA levels of ATG4A, AMBRA1, and ULK1. Furthermore, miR-204-3p regulates the initiation of autophagy through ULK1-dependent activation of the VPS34 complex, and inhibition of ULK1 eliminated the increase in Beclin1 phosphorylation and VPS34 activity.

Fig. 5. miR-204-3p inhibits macrophage inflammation by restoring ULK1-dependent autophagy.
To assess the functional relationship between miR-204-3p and ULK1 in hepatic steatosis, the authors injected AAV-control or AAV-204-3p into HFD-fed Ulk1-/- mice. They found that Ulk1 deletion led to increased liver weight/body weight ratios and that macrophage-specific miR-204-3p upregulation could not reverse this condition. Additionally, Ulk1-/- mice had decreased PparamRNA levels and increased levels of pro-inflammatory factors and inflammatory signaling molecules in the liver. These results indicate that the protective effect of miR-204-3p on liver fat accumulation is primarily dependent on ULK1.

Fig. 6. Ulk1 deficiency abolishes the protective effects of miR-204-3p on MASLD.
Conclusion
This study reveals a novel function of macrophage miR-204-3p, which acts through the transcriptional regulation of ULK1 expression. It enhances autophagic flux, reduces inflammation, and thereby limits fatty liver disease. Consequently, miR-204-3p and potential specific ULK1 agonists could serve as new therapeutic targets for metabolic dysfunction-associated fatty liver disease (MASLD).








